大阪市立大学(大阪市大)は9月2日、量子コンピュータを用いて原子や分子の任意のエネルギー差を直接計算できる「量子位相差推定」アルゴリズムを開発したと発表した。
同成果は、大阪市大大学院 理学研究科の杉﨑研司特任講師、同・佐藤和信教授、工位武治名誉教授らの研究チームによるもの。詳細は、物理化学、化学物理学、生物物理学を扱う学術誌「Physical Chemistry Chemical Physics」に掲載された。
量子コンピュータの近い将来の計算ターゲットとして注目されているのが、量子力学の基礎となる「シュレーディンガー方程式」を近似的に解き、電子状態を明らかにする「量子化学計算」だという。
「全配置間相互作用法」(full-CI法)と呼ばれる精密な量子化学計算は、波動関数展開において、可能なすべての電子配置を考慮する方法だが、従来のコンピュータでは分子サイズに対して指数関数的に計算コストが増大してしまうことが課題とされ、量子コンピュータでの活用が期待されてきたが、量子コンピュータを用いた量子化学計算であっても、エネルギー計算値の誤差に反比例して計算コストが増えるため、全エネルギーを小さな桁まで正確に求めるには大きな労力を要してしまうという。
その一方で、可視光などの電磁波の吸収波長を計算して分子を同定する、化学反応の進行に必要なエネルギー障壁の高さを求めるなど、量子化学計算で扱う問題のほとんどは分子の全エネルギーそのものではなく、エネルギー差を議論するほか、電子励起エネルギーやイオン化エネルギーなど、化学で議論されるエネルギー差の大きさは分子サイズにあまり依存しないという特徴があることから、研究チームでは、全エネルギーではなくエネルギー差を量子コンピュータで直接計算することができれば、量子コンピュータを実際の化学研究や物質開発に役立てられると考え、研究を進めてきたという。
量子コンピュータによるfull-CI計算に用いられる量子位相推定アルゴリズムは、波動関数(Ψ)を時間発展させると、全エネルギーに依存した速さで波動関数の位相が変化する事象を利用した量子アルゴリズムであり、今回の研究では、この量子位相推定アルゴリズムを応用し、エネルギー差を求めたい2つの電子状態の波動関数の量子重ね合わせ状態を準備し、その量子状態を時間発展させることで、時間発展後2つの電子状態の波動関数の位相差(エネルギー差)を直接計算できる「量子位相差推定」アルゴリズムを開発することに成功したという。
-
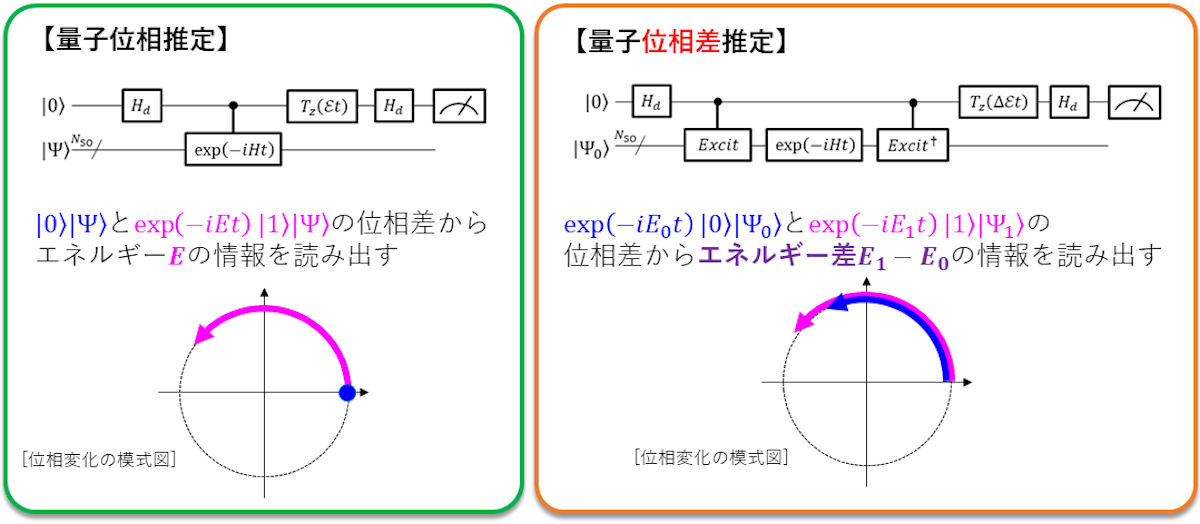
(左)従来法である量子位相推定アルゴリズムの概要。(右)今回の研究で開発された量子位相差推定アルゴリズムの概要 (出所:大阪市大プレスリリースPDF)

すでに研究チームはスピン量子数が異なる電子状態(スピン状態)間のエネルギー差を直接計算する量子アルゴリズムを開発済みだが、この量子アルゴリズムは、実装に必要な量子ビット数が、従来法である量子位相推定よりも増えてしまうという点と、紫外-可視吸収スペクトルの帰属などで重要となる、スピン量子数が等しい電子状態間の励起エネルギーを求めることができないという2つの課題があったという。これに対し、今回開発された量子位相差推定アルゴリズムはこれらの課題を解決していることから、汎用性の高い量子アルゴリズムになったと説明している。
なお量子位相差推定アルゴリズムは、量子化学計算以外にも適用できることから、さまざまな物理問題・数学問題への応用が期待されると研究チームでは説明しているほか、従来法の量子位相推定では必須だった制御-時間発展演算が不要となるため、量子コンピュータ実機への実装が量子位相推定よりも容易になることが期待されるともしている。