東京大学(東大)は4月26日、「超並列ミトコンドリアゲノム(ミトゲノム)シーケンス」を、ハゼ科の魚類である「アゴハゼ」の日本海系統に適用することで、「分子系統地理学」における懸念であった「ランダムルーティング問題」を解消することに成功したと発表。さらに、ルーティングの安定した信頼性の高い分子系統樹を得ることにより、新たな地域系統の発見や正確な分岐年代推定など、分子系統地理学において重要な知見が得られることを示したとした。
成果は、同大 生物科学専攻 特別研究員兼東大大学院 農学生命科学研究科の平瀬祥太朗 助教(当時)らの研究チームによるもの。研究の詳細な内容は、4月28日付けで「Genome Biology and Evolution」に掲載された。
分子系統地理学とは、近縁な生物集団間の空間的・時間的な関係性を、DNAなどを用いて明らかにし、それらの分布域形成の歴史を明らかにする学問分野であり、同一種でも地域の集団間で系統の分岐などが生じていた場合、それらの地域間に何らかの地理的障壁があり移動分散が制限されていた可能性があることなどを知ることができる。
生物集団が過去の地理的イベントの影響などを受けつつ、時間軸に沿ってどのように拡大や移動を行ってきたかを解明する上では、対象としている生物集団に含まれない近縁関係にある外群の情報を用いて分子系統樹の根を決定する「ルーティング」を正確に行うことが重要だという。しかし、ルーティングは一般の進化学的解析においても決して簡単な作業ではなく、中でも分子系統地理学においては、ごく近縁な集団の遺伝的関係の解析を行うために、対象としている生物集団内におけるDNA配列の変異についての時間軸的な情報を得ることが難しいという課題があり、結果として、根の位置が決定できないという困難が生じてしまうという「ランダムルーティング問題」が起こっていた。
従来、分子系統地理学ではコストや時間の制約から「ミトコンドリアDNA」の部分配列を解析対象する手法が用いられてきたが、今回の研究では、新型のDNAシーケンサーを用いて多数の個体におけるミトコンドリアDNAの全塩基配列(ミトコンドリアゲノム:ミトゲノム)情報を一挙に、高速かつ低コストで得ることで、これまで得ることが困難だった大規模データに基づいた解析を可能にしたという。
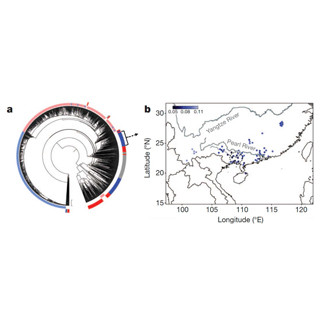
今回、ランダムルーティングのモデルケースとして更新世(約258万8000年前~1万1700年前)の日本海隔離によって生じ、その後、近年になって急激に集団拡大したアゴハゼの日本海系統を解析対象として解析を実施。具体的には、日本列島と朝鮮半島沿岸の18地点で採集を行い、超並列ミトゲノムシーケンスを実施。解析の結果、ルーティングの安定した信頼性の高い分子系統樹が得られたとする(画像1)。

|
|
画像1 超並列ミトゲノムシーケンスによるアゴハゼの分子系統樹。従来のミトコンドリアDNAの部分配列のみの解析だと、ランダムルーティング問題が生じていたという |
また、これまで知られていなかった日本海系統内における北部系統と南部系統の存在も判明。中でも北部系統が近年になって急激な集団拡大を経験し、南部系統はそうした拡大を経験しなかったことも示唆されたとしている。
なお研究チームでは、北部系統の共通祖先年代を較正点として利用することで、日本海系統と太平洋系統の分岐年代をより正確に推定することも可能となり、日本海の環境を大きく変えた過去の地史イベントがアゴハゼに与えた影響を考察することができるようになったとする。具体的には、およそ350万年前の対馬海峡が開いた時期に日本海に侵入し、それ以降の隔離によって、太平洋側と日本海側の集団が分断されたことが考えられるとしている。