理化学研究所(理研)は5月10日、北海道大学、横浜市立大学、骨系統疾患コンソーシアム所属の日本全国の臨床医、埼玉県立小児医療センターとの共同研究により、骨や関節、軟骨、靱帯、皮膚など広い範囲の組織で異常を引き起こす一連の遺伝性難治疾患の原因が、「グリコサミノグリカン(GAG)」という糖鎖の合成に重要な「B3GALT6遺伝子」であることを発見したと発表した。
成果は、理研 統合生命医科学研究センター 骨関節疾患研究チームの池川志郎チームリーダー、同・中島正宏特別研究員、北大大学院 先端生命科学研究院の菅原一幸教授、同・水本秀二博士研究員、横浜市立大 環境分子医科学の三宅紀子准教授、同・松本直通教授らの研究チームと、骨系統疾患コンソーシアム所属の日本全国の臨床医、埼玉県立小児医療センター・遺伝科の大橋博文部長の運営する細胞・DNAバンクなどによるものだ。
骨・関節には数多くの遺伝性難治疾患が存在する。その中の1つである「関節弛緩を伴う脊椎骨端骨幹端異形成症I型」(SEMD-JL1:spondyloepimetaphyseal dysplasia with joint laxity, type1)は、脊柱が後方や側方へねじれ曲がる後側彎などの脊椎の変形や、肘や膝の関節の変形や脱臼など、重度の骨格異常を来す難病だ(画像1・A~C)。

|
|
画像1。関節弛緩を伴うSEMD-JL1(34歳、男性)。(A):外観。短体幹型(胴体が短くなるタイプ)の低身長(118cm)、突き出した胸骨、肘の変形が見られる。顔貌、筋緊張の異常はない。(B):脊椎側面のX線像。脊椎の発達障害による重度の後側彎。(C):股関節正面のX線像。腸骨の短縮、大腿骨小転子の突出、大腿骨頭の短縮が見られる |
SEMD-JL1は「常染色体劣性の遺伝形式」を採る「単一遺伝子病」であることは知られている遺伝病だ。常染色体劣性の遺伝形式とは、メンデル式の遺伝形式の1つで、常染色体に存在する、ある1つの遺伝子の異常によって起きる疾患の遺伝形式をいう。父母、それぞれから由来する1対(2コピー)の遺伝子の内、両方のコピーに異常があった時に疾患が発症する。
そして単一遺伝子病とは、1つの遺伝子の異常によって起きる病気のこと。メンデル式の遺伝形式を採る。これに対し、2種類以上の異常遺伝子と環境因子が関与して起きる病気を「多因子遺伝病」という。通常、遺伝病というと単一遺伝子病を指す。つまり、SEMD-JL1とは、父母両方からもらった1つの遺伝子がどちらも異常があるために起きてしまう疾患ということである。ただし、その肝心の異常を来している原因遺伝子そのものがこれまでのところ発見されていない。
またSEMD-JL1は現在、臨床症状やX線画像に基づいて診断されているため、別の骨・関節疾患と誤診されたり、診断がつけられなかったりすることもあり、アジアでは1例も見つかっていなかった。この疾患の重度の骨格異常は大きな手術を必要とすることが多く、治療が困難なため、早期の正確な診断と治療が重要である。
しかし、ここに来て次世代シーケンサー技術が進歩してきたことにより、難治疾患患者の遺伝子解析を網羅的かつ極めて迅速に行うことが可能となってきた。そこで研究チームは、次世代シーケンサーを用いた大規模ゲノム解析の手法を用いて、この疾患の原因遺伝子の解明に挑んだというわけだ。
研究チームは、SEMD-JL1と診断できる症例として、6家系(日本人5家系と日本人とシンガポール人の1家系)7例の検体を収集し、次世代シーケンサーで「エクソーム解析」を実施。エクソーム解析とは、ゲノムの中のタンパク質に関する情報を含むエクソン部分(ゲノム全体の約3%)を、次世代シーケンサーを用いて包括的に解析する方法のことだ。その結果、すべての症例に「B3GALT6遺伝子」の変異が発見された(画像2の表の家系ID1~6)。
それらの変異はすべて「B3GALT6タンパク質」の機能を喪失させる変異と推測され、7例中6例はB3GALT6遺伝子の「複合ヘテロ接合性変異」だった。複合ヘテロ接合性変異とは、大まかにいうと、遺伝的な変異を持っているが発症していない保因者(変異を持っているのに発症していない人。本人は遺伝病ではないが、子供が遺伝病を発症する可能性を持つことになる)が両親の時、両親それぞれから異なる変異を伝達された状態のことをいうのである。
もう少し詳しく述べると、個体は卵と精子を通して1セットずつのゲノムの計2セット持つため、遺伝子には父母由来の2つの対立遺伝子が存在し、その特定の対立遺伝子の組み合わせが異なる場合を「ヘテロ接合(異型接合)」という。疾患の原因となる変異を有する遺伝子において、保因者である父親と母親の双方から異なる変異を伝達された(継承した)状態を特に複合ヘテロ接合(compound heterozygote)というのである。
さらに、ベトナム人の1家系(画像2の表の家系ID7)でもB3GALT6遺伝子の複合ヘテロ接合性変異を見つけた。その変異の内の1つ「694C>T」は、日本人患者3家系(画像2の表の家系ID1、4、5)で見つかったものと同じだったことから、この変異は東アジアに広く分布していることが示唆されるという。
画像2の表が、関節弛緩を伴うSEMD-JL1と先天性結合組織疾患の1つである「エーラス・ダンロス(Ehlers-Danlos)症候群早老性型(EDS-PF)」患者で発見したB3GALT6遺伝子の変異。項目「DNAの塩基の変化」の見方だが、例えば694C>Tの場合、「B3GALT6遺伝子における694番目の塩基であるシトシン(C)がチミン(T)に変わる変異」という意味だ。「予想されるアミノ酸の変化」の見方は、例えば「Arg232Cys」の場合、「B3GALT6タンパク質の232番目のアミノ酸Arg(アルギニン)が Cys(システイン)に変わる変異」ということになる。

|
|
画像2。関節弛緩を伴う脊椎骨端骨幹端異形成症I型(SEMD-JL1)と先天性結合組織疾患の1つである「エーラス・ダンロス症候群早老性型(EDS-PF)」患者で発見したB3GALT6遺伝子の変異 |
研究チームは次に、EDS-PFについてもB3GALT6遺伝子が調べられた。この疾患の主な症状は、特徴的な顔貌、靱帯の弛緩性、皮膚過伸展、筋緊張の低下だ(画像3・D~G)。脊椎や関節の異常も引き起こすが、これまではSEMD-JL1とはまったく別の疾患として考えられていた遺伝病である。

|

|
|
EDS-PFの臨床像(5歳、女児)。画像3(左)の(D):外観。特徴的な顔貌(平らな顔、突き出した目、垂れ下がった頬を伴う顔面中心部の低形成)、しわの多い皮膚、筋緊張の低下、肘の変形、内反尖足(足の底が内側を向き(内反)、足首が伸びる(尖足))が見られる。(E):脊椎側面のX線像。胸椎および腰椎の後彎、扁平で前方の先端が尖った椎体などの特徴が見られる。画像4(右)の(F)、(G):肘関節と股関節正面のX線像。肘および左股関節の脱臼、長管骨の骨端および骨幹端の形成異常が見られる |
|
今回B3GALT6遺伝子の変異を見つけたSEMD-JL1の7家系(画像2の表の家系ID1~7)8例の臨床像とX線像が詳細に検討された結果、EDS-PFの病像とSEMD-JL1とに多くの重複があることが判明。そこで、EDS-PFの患者3家系4例を対象にB3GALT6遺伝子を解析した結果、すべての検体からSEMD-JL1と同様にB3GALT6遺伝子の複合ヘテロ接合性変異が発見された。SEMD-JL1と同様に、その変異はB3GALT6タンパク質の機能を喪失すると推測できた(画像2の表の家系ID8~10)。
B3GALT6タンパク質は、グリコサミノグリカン(GAG)の結合領域の糖転移酵素の1つであり、「プロテオグリカン(PG)」の合成に重要な役割を果たす酵素として知られている。患者由来の細胞を用いて詳しい調査が行われたところ、今回発見した変異は、2つのタイプの異常によりB3GALT6タンパク質の機能を喪失させる可能性が示された。
1つは細胞内でのB3GALT6タンパク質の分布に異常を引き起こすタイプだ。GAG結合領域の合成は細胞内小器官の「ゴルジ体」(細胞外へ分泌されるタンパク質の糖鎖修飾、切断、貯蔵などの機能を持つ)で行われるため、B3GALT6タンパク質も本来ゴルジ体に存在している。
しかし、変異型の場合はゴルジ体以外の部位にB3GALT6タンパク質が分布するため、酵素機能は正常に発揮できないと考えられた(画像5)。もう1つは、糖転移酵素としての機能自体が喪失するタイプだ(画像6)。患者由来の細胞を用いたGAGの生化学的解析が行われた結果、GAGの1種で、生体内のさまざまな分子と相互作用する「ヘパラン硫酸」が、健常人に比べて26~56%減少していることが突き止められた。
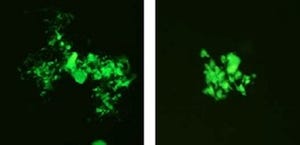
画像5と画像6は、SEMD-JL1で発見したB3GALT6遺伝子変異によって起こる異常。画像5は、蛍光免疫染色法で調べた変異体(Met1?)の細胞内局在。正常型の場合、B3GALT6タンパク質は核の周囲に分布し、ゴルジ体の指標タンパク質である「α-マンノシダーゼ」と重なるのでB3GALT6タンパク質がゴルジ体に存在するのがわかる。それに対して変異型の場合は、B3GALT6タンパク質はゴルジタイ以外の部位に拡散して分布しα-マンノシダーゼと重ならないので、ゴルジ体に存在していない。
画像6は、変異型タンパク質の糖転移活性。変異型タンパク質では正常型に比べて糖転移活性が非常に低い。以上の結果から、SEMD-JL1患者ではB3GALT6タンパク質の局在の異常、または酵素活性の低下により、機能に障害があると考えられた。

|

|
|
SEMD-JL1で発見したB3GALT6遺伝子変異によって起こる異常。画像5(左):蛍光免疫染色法で調べた変異体(Met1?)の細胞内局在。画像6(右):変異型タンパク質の糖転移活性。対照とはB3GALT6遺伝子が導入されていない場合。*有意水準 P<0.0001(vs.正常型) |
|
このように、B3GALT6タンパク質の機能障害でGAG結合領域が正常に合成されないと、皮膚過伸展、筋緊張の低下、靱帯や関節の可動性亢進、および脊椎の変形など広範な骨格、結合組織の異常を引き起こすことがわかったのである。
近年、GAG結合領域の合成に関わる「B4GALT7」や「B3GAT3」など、ほかの酵素の機能障害でも、SEMD-JL1やEDS-PFに似た一連の病像を示すことが相次いで報告されている。つまり、B3GALT6を初めとするGAG結合領域の合成酵素は、多様なヒトの組織の形成や維持において重要な役割を担っていることが明らかになったというわけだ。これらの疾患群は、「GAG結合領域病」として統一した理解が可能だと考えられるという。
B3GALT6遺伝子の発見は、従来診断が困難だった一連の骨格・結合組織の異常を引き起こす難病の遺伝子診断、確定診断、保因者診断を可能にする。研究チームによれば、今回の結果は、診断されていない多数のSEMD-JL1患者が東アジアに存在することを示唆しているという。ただし、今後は遺伝子診断が普及していけば、SEMD-JL1やEDS-PFなど「GAG結合領域病」患者の早期発見、そして治療が可能になるとしている。
さらにB3GALT6遺伝子の解析は、SEMD-JL1疾患での側彎や関節脱臼の発症のメカニズム解明だけでなく、一般の側彎や関節脱臼のメカニズム解明や治療にも大きな知見をもたらすと期待できるいう。研究チームは今後、B3GALT6タンパク質の機能解析を通じてGAG結合領域病の病態を解明し、酵素の補充療法など画期的な治療法の開発を目指すとした。