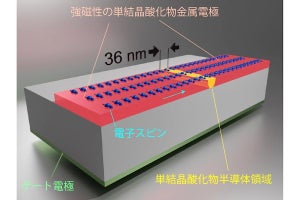
東京大学(東大)は11月15日、ガリウム(Ga)・マンガン(Mn)・ヒ素(As)からなる強磁性半導体の代表的な存在である「(Ga,Mn)As」において、電気伝導特性が特異な振る舞いを示す原因を、改良した「第一原理計算」により解明することに成功したと発表した。
-

温度効果による電子散乱の概念図(出所:東大プレスリリースPDF)

同成果は、東大大学院 工学系研究科附属 スピントロニクス学術連携研究教育センターの新屋ひかり特任准教授(電気系工学専攻兼任)、同・大矢忍教授(電気系工学専攻兼任)、同・吉田博嘱託研究員(大阪大学 名誉教授兼任)、産業技術総合研究所 機能材料コンピュテーショナルデザイン研究センターの福島鉄也研究チーム長、大阪大大学院 工学研究科の佐藤和則准教授らの共同研究チームによるもの。詳細は、米国物理学協会が刊行する材料科学を扱う学術誌「APL Materials」に掲載された。
新しい高機能デバイスにふさわしい物質を発見するためには、動作温度における電気伝導特性が重要な指標となる。強磁性半導体の(Ga,Mn)Asでは、低温域では温度が上昇すると電気抵抗率が増大し、高温域では温度の上昇に伴い電気抵抗率が減少する。一般的に、金属の電気抵抗率は温度が上昇すると増大するが、半導体では温度の上昇に伴い電気抵抗率が減少するので、(Ga,Mn)Asにおける電気抵抗率の振る舞いは非常に特異な現象だという。この特異な振る舞いの原因は未解明だが、電気伝導特性を左右する要因を明らかにすることは、強磁性半導体の応用開発を行うにあたり重要なミッションだとする。
未解明の物理現象の謎を解き明かすには、現在では理論的なシミュレーションは欠かせない。中でも第一原理計算は、コンピュータ上のプログラムに結晶構造を与えるだけで物質の特性をシミュレートできることから、既存の物質だけでなく未知の物質空間も手広く調べることが可能で、実に数多くの研究で用いられている。
第一原理計算は単なる理論シミュレーションというわけではなく、量子力学に基づいた確かな手法であり、産業界でも活用されている。しかし同手法は、絶対零度における理論である「密度汎関数法」に基づいた手法であるため、有限温度における物理量を計算するためには、温度の効果を取り入れることが必要だという。また、電気伝導特性も密度汎関数法の範疇では計算することは困難とする。
そうした中でこれまで、従来の第一原理計算における上述した2つの課題を克服するため、「有限温度における電気伝導特性が計算可能な新しい第一原理計算手法」の開発に取り組んできたのが研究チームだ。強磁性半導体中では温度の上昇により原子振動やスピンゆらぎが生じるが、これらによって電子が散乱される効果を考慮すれば、有限温度における物質の特性を計算することが可能となるという。
今回の研究ではこのような温度効果について、「コヒーレントポテンシャル近似」(CPA)と呼ばれる手法を利用し、第一原理計算に取り込んだとのこと。また、線形応答理論を利用することで、温度効果を取り込んだ状態かつ経験的な情報を用いることなく、電気伝導特性を計算することができるとしている。
さらに今回の研究では、この新しい第一原理計算手法を利用して、(Ga,Mn)Asの電気伝導特性が示す特異な振る舞いの原因の解明に取り組んだという。(Ga,Mn)Asの結晶内部では、Ga原子の位置を置き換えたMn原子(置換型原子)と、本来の原子位置の隙間に侵入するMn原子(格子間原子)が自然発生するが、今回の研究により、これらの2種類のMn原子が電気伝導特性に大きな影響を与えていることが明らかにされた。特に低温域では、格子間Mn原子において生じるスピンのゆらぎが、電気伝導を大きく妨げる役割を果たしていることが判明した一方、高温域では電気伝導を主に担っているAs原子の電子状態が原子振動により変化することで、抵抗が小さくなっていることがわかったとする。
-

半導体GaAsと強磁性半導体(Ga,Mn)Asの結晶構造(出所:東大プレスリリースPDF)

-

今回の研究による温度‐電気抵抗率の計算結果(出所:東大プレスリリースPDF)

研究チームは、今回開発された新たな手法が強磁性半導体以外の材料系にも適用することが可能であり、あらゆる分野において、材料開発の時間短縮や低コスト化に貢献することが期待されるとしている。