生理学研究所は、心筋細胞膜で高血糖時に発現増加するCa2+透過型カチオンチャネル(transient receptor potential canonical:TRPC)6チャネルが、心筋細胞での活性酸素の生成を抑制することで心不全発症リスクを軽減することをマウスを用いて明らかにしたと発表した。
同研究は、生理学研究所心循環シグナル研究部門の西田基宏教授(九州大学教授兼務)、総合研究大学院大学・大学院生の小田紗矢香の研究グループと、味の素製薬(現EAファーマ)、筑波大学、米国国立環境健康科学研究所(NIEHS)との共同研究よるもので、同研究成果は、日本時間8月8日、オンライン科学雑誌であるScientific Reports誌に掲載された。

|
インスリンの分泌を阻害する薬剤によって高血糖状態にした「TRPC6チャネルを欠損させたマウス」では、野生型マウス(WT)やTRPC3欠損マウスと比べて突然死する個体が増加したり、全身へ血液を送る左心室の筋力が衰え、顕著な収縮力の低下をきたした。また、マウス心臓を高血糖状態にすることでTRPC6チャネルの発現量が増加する一方、活性酸素生成酵素(Nox2)の発現量は低下することも明らかになった。 (出所:生理学研究所プレスリリース) |
糖尿病による心機能低下は、冠動脈硬化を伴う虚血性心疾患や高血圧とは独立した心病変として臨床的に注目されており、その原因として心筋での酸化ストレス障害が示唆されていたが、発生機序は不明だった。
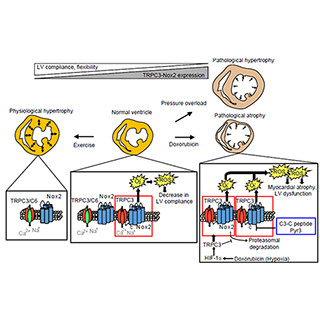
2016年、西田教授の研究グループは、心筋細胞膜上に存在するTRPC3チャネルがNADPHオキシダーゼ2(Nox2:酸化ストレスの原因となる活性酸素を生成する酵素)と相互作用することで、Nox2タンパク質自体が分解されるのを抑制するとともに、Nox2自体の安定化にも寄与していることを報告した。さらにTRPC3チャネルは、心筋の伸び縮みによって活性化することから、Nox2からの活性酸素生成を促し、心臓の線維化(硬化)を誘導する、という心筋梗塞のメカニズムの一端を明らかにした。
今回の研究では、TRPC3チャネルと同じく心筋細胞に存在し、かつよく似た構造・機能を持つTRPC6チャネルというタンパク質に注目。まず最初に研究グループは、高血糖状態のマウスの心臓とラットの心筋細胞ではTRPC6チャネルタンパク質の量が増加し、TRPC3チャネルとNox2が相互作用をしないよう阻害していることを明らかにした。つまりTRPC6チャネルは、活性酸素の生成を抑制し、心臓を保護するよう働いているということを意味している。

|
高血糖負荷により心臓で適切にTRPC6発現量を増加させることがTRPC3-Nox2複合体形成による酸化ストレス発生を抑制し、糖尿病性心不全発症のリスク低下につながる可能性が示された。(出所:生理学研究所プレスリリース) |
加えてインスリン分泌機能を止める薬剤で「野生型マウス(正常マウス)」と「TRPC3チャネルが欠損したマウス」、「TRPC6チャネルが欠損したマウス」といった3種類のマウスをそれぞれ高血糖状態にしたところ、TRPC6チャネルを欠損したマウスのみが生存率や心機能の低下をきたし、酸化ストレスが顕著に増加した。また、高血糖状態になった野生型マウスの心臓やラット胎児の心筋細胞ではTRPC6チャネルの発現量が増加し、Nox2の発現量は低下していた。
さらにTRPC3チャネル、TRPC6チャネル、Nox2のそれぞれを過剰に発現させた細胞を用いた実験の結果、TRPC6チャネルはTRPC3チャネルと複合体を形成することでTRPC3とNox2が複合体を形成するのを抑制し、結果的にNox2の発現量を低下させていることが明らかになった。つまり高血糖状態の心筋でTRPC6チャネルが増加することは、結果として心筋の酸化ストレスを軽減し、心臓を保護する役割につながっていることを示唆している。
糖尿病性心不全は突然死リスクの高い糖尿病合併症の一つであり、その予防・治療法の開発は極めて重要となる。高血糖負荷により心臓でTRPC6タンパク質が適切に発現増加できる機構を明らかにし、これを維持あるいは促進する方法を開発することで、糖尿病性心不全の発症リスクを軽減することが期待されるということだ。