名古屋工業大学(名工大)は8月21日、全固体リチウム電池(LIB)材料で重要な電極・電解質界面について、「深層学習ポテンシャル」(以下、DLP)を用いた分子動力学シミュレーションを行い、原子レベルでの固体電解質の化学的安定性、界面における「空間電荷層」の形成、界面リチウムイオン(以下、Li+)交換反応速度の定量化などの評価に成功したことを発表した。
同成果は、名工大 工学専攻創造工学プログラムの岩崎梨音大学院生(研究当時)、同・大学 生命・応用化学類の中山将伸教授らの研究チームによるもの。詳細は、英科学誌「Nature」系の材料科学に関する全般を含めた学術誌「Communications Materials」に掲載された。
全固体LIBは、正極、負極、電解質などの主要電極部材がすべて固体であることから、接触界面におけるLi+の界面交換反応を適切に制御する必要が求められている。これまでのところ、界面に電気抵抗が発生しており、エネルギー変換のロスや出力特性の低下につながることが明らかにされている。また、接触した2つの固体間での分解反応により、電池が劣化する現象なども観測されてきた。しかし、原子レベルでどのような反応が発生しているのかは未解明のままだとする。
量子力学計算を用いた材料シミュレーションによる反応機構の解明が進められているが、計算負荷が大きく、入力可能なモデルサイズの制限や、計算リソースなどに対する経済的コストなどの課題が生じているという。そこで研究チームは今回、従来用いられてきた量子力学計算に代わり、DLP計算を用いて材料シミュレーションを行うことにしたとする。
-
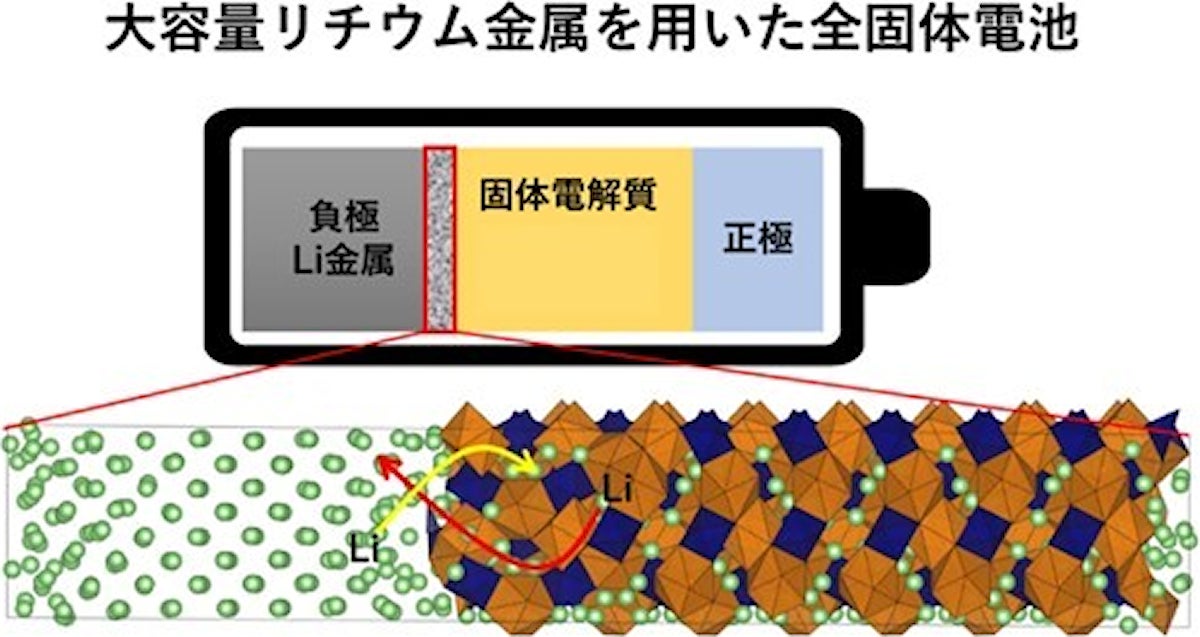
シミュレーションで可視化する界面ナノスケール空間とリチウムの交換反応(出所:名工大Webサイト)

DLPとは、量子力学計算の結果データベースをあらかじめ学習することで、ユーザーが与えた結晶構造のエネルギーや原子に作用する力を即時に評価できるという手法論。同手法を用いて、金属Li負極とガーネット型固体電解質「Li7La3Zr2O12」(以下、「LLZ」)の接触界面のシミュレーションが行われた。これまでに行われた量子力学計算(DFT計算)によるLi/LLZ界面の研究では298原子による評価だったが、今回は1024原子に拡張し、さらにDFT計算では困難だったLi+の移動過程についての検討も行うことにしたという。
-
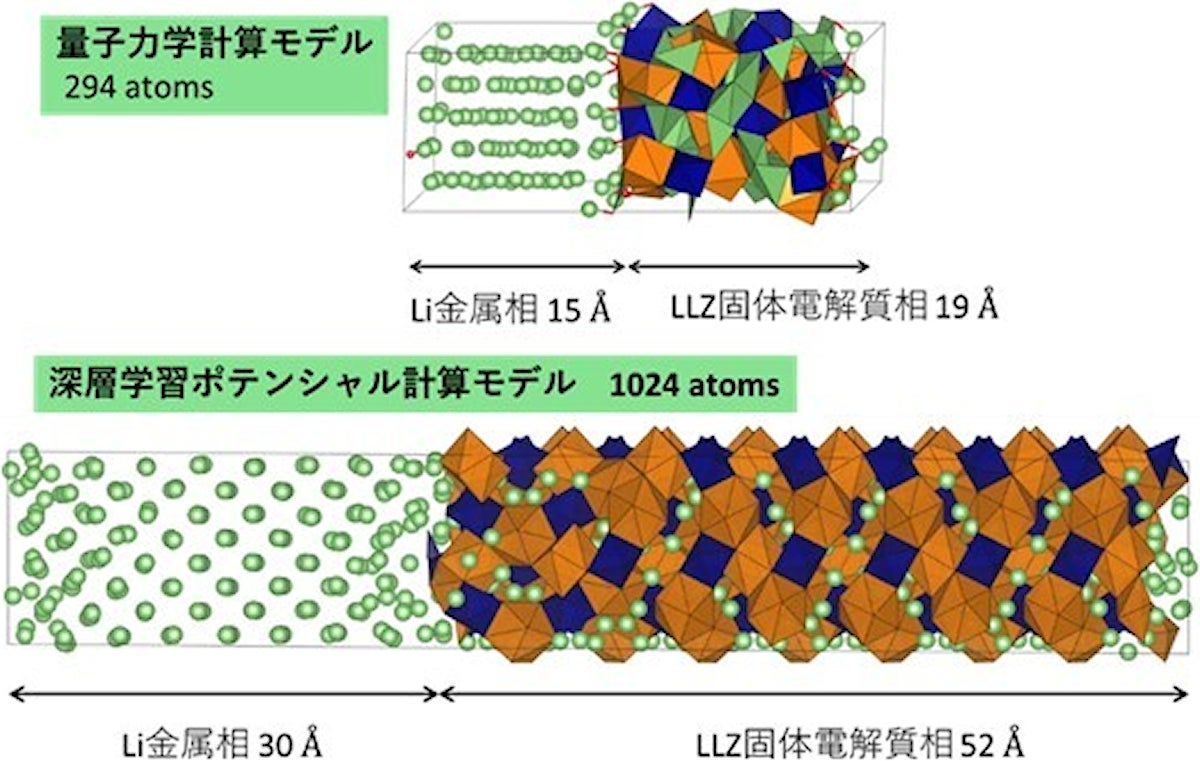
これまでの量子力学計算に使用されたモデルと、今回の研究で用いられたDLP計算に使用されたモデル。DLPを用いると計算が高速化されるため、固体電解質を十分厚くした状況で評価が可能となる(出所:名工大Webサイト)

結果として、LLZは高温(1000℃)でも金属Liと分解反応する挙動は観測されず、非常に安定な固体電解質材料であることが確認されたとする。また、Li+の移動過程が時系列で解析された結果、固体電解質の表面に正電荷を有するLi+がLLZよりも過剰に堆積した状態が形成されることが判明した。LLZにて正電荷が過剰に堆積した空間を形成した結果、一定空間に電荷が溜まった領域である空間電荷層が形成されたものと考えられるという。
-
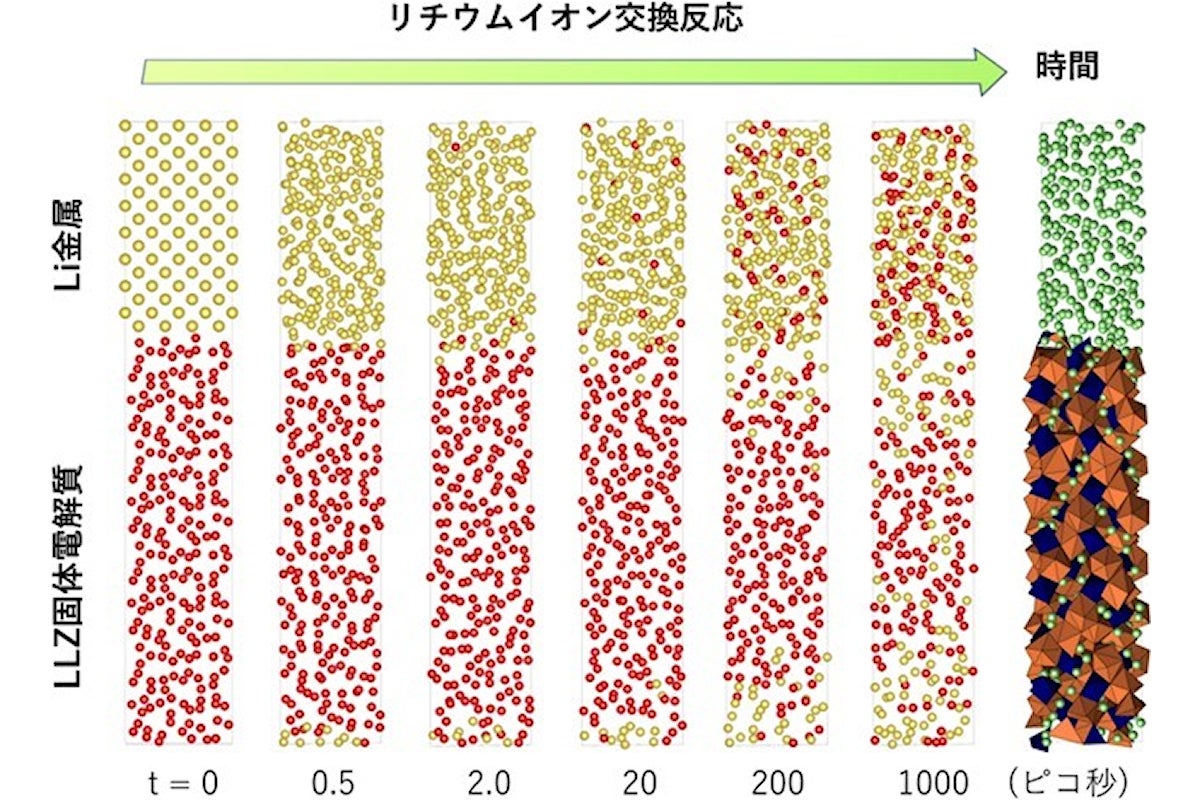
LLZと金属Liの間で移動するLiを時系列に可視化した図。時刻0の時に、金属Li相にあるLiが黄色、LLZ相にあるLiが赤色で表されており、時間が経過するにつれて、2つの相のLi+が混ざり合っていくことがわかる(出所:名工大Webサイト)

DLPによる1ナノ秒の分子動力学計算の結果を用いて、電子構造が可視化され、Li/LLZ界面近傍で、LLZ相のバンドが1.1ナノメートルにわたって折れ曲がっているという結果が得られ、空間電荷層の存在が確認されたとした。同層の形成は、Li金属からLLZ相に電子が移動し還元分解を抑制するため、固体電解質の安定性を向上させることにつながるとする。
-
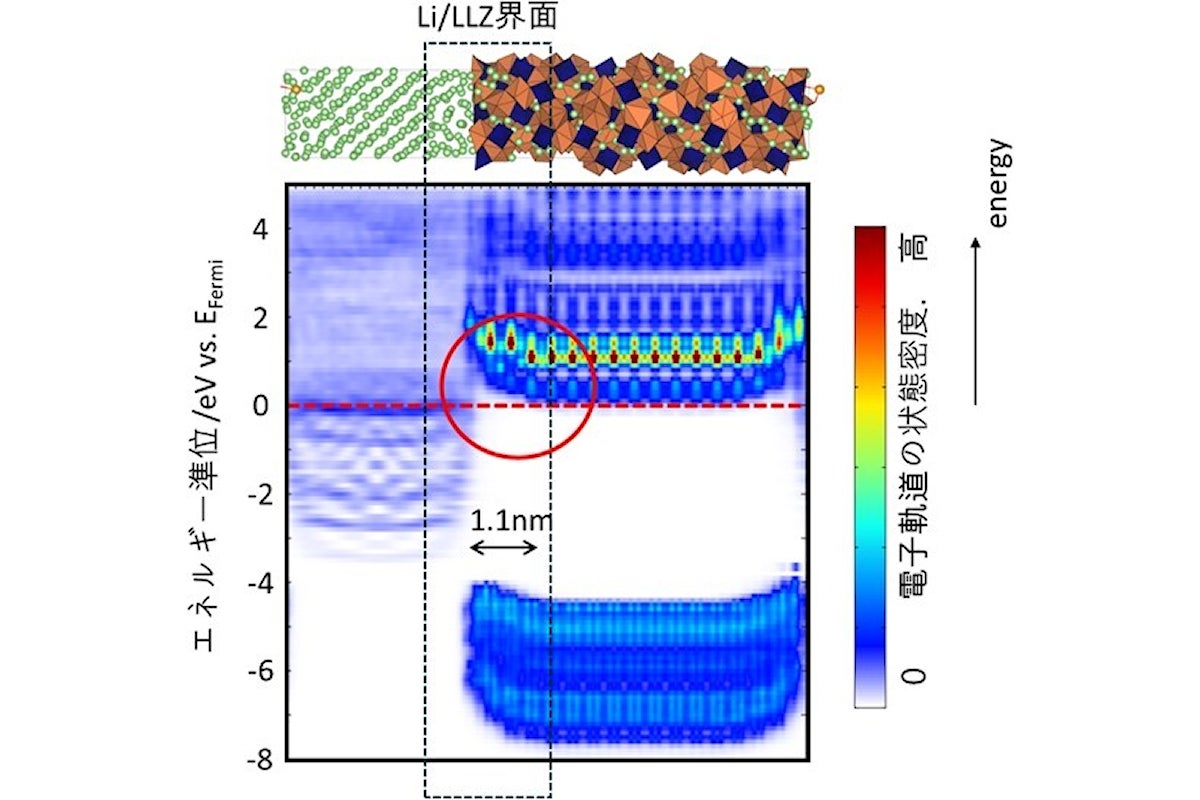
DLPで得られたLi/LLZ界面モデル構造を用いて、量子力学計算により再計算を1回行い、電子状態解析を行った結果。Li/LLZ界面近傍(黒破線枠)において、赤丸で囲んだLLZ相の伝導帯バンドが屈曲しており、空間電荷層が形成されたことが示されたとした(出所:名工大Webサイト)

さらに、界面におけるLi+交換反応速度もシミュレーションで定量化することに成功したという。LLZ相からLi相、Li相からLLZ相へのLi+の移動の活性化障壁エネルギーは160ミリ電子ボルト(meV)および88meVであり、非常に低い値が示されたとした。界面でのLi+の交換は、Li/LLZ界面では非常にスムーズであり、固体内のイオンの拡散が律速していることが定量的に確認されたとする。
今回の研究成果はLi/LLZ界面にとどまらず、多くの電池材料に当てはめることができるという。また、実験で得られる複雑なスペクトルの解釈に悩むことなく、直観的に界面で発生している反応も可視化でき、全固体LIBの設計のための研究を加速させることが期待されるとした。
また今回の成果は、負極と固体電解質のみではなく、正極と固体電解質の反応性評価にも用いることが可能だという。また、電極と固体電解質間ではしばしば副反応が発生し、コート材を導入するなどのプロセスが必要なことが経験的に知られているが、原子レベルでコート材がどのような役割を果たしているのかは、いまだに不明な点が多い。全固体LIBで経験的に用いられている技術やノウハウについても、体系的解明に近づくことが期待されるとした。体系的な界面設計指針が得られることで、膨大な組み合わせが存在する電極・電解質・コート材などの選定をシミュレーションやインフォマティクスで行うことが可能となり、電気自動車電源やスマートグリッド用電源となる安全かつ大容量全固体LIBの実現に結びつくとしている。