横浜市立大学は12月10日、脳性麻痺やてんかん、精神遅滞などを引き起こす脳奇形「孔脳症」および「裂脳症」の患者の約2割に、「IV型コラーゲンα1鎖」をコードする遺伝子「COL4A1」の「ヘテロ接合性変異」があることを同定したと発表した。
成果は、横浜市大大学院 医学研究科の米田祐梨子氏、同・学術院医学群の才津浩智准教授(遺伝学教室)を中心とした、宮城県拓桃医療療育センターの萩野谷和裕副院長、山形大学小児科の加藤光広講師、神奈川県立こども医療センターの小坂仁部長らの共同研究グループによるもの。研究の詳細な内容は、日本時間12月8日付けで米国小児神経学会雑誌「Annals of Neurology」オンライン版に掲載された。
孔脳症は大脳半球内に「脳室」との交通を有する「嚢胞(のうほう)」または空洞が見られる脳奇形で、胎生期における梗塞(こうそく)や出血といった脳循環障害により発生すると推測されている。海外での発症率は10万人に0.5~3.5人程度とされているが、日本での正確な頻度は不明だ。
また、裂脳症は脳室から大脳半球表面まで達する裂溝が存在し、その表面が異常灰白質で覆われる脳奇形である。神経細胞の遊走異常を伴う点が孔脳症と大きく異なるが、孔脳症と同様に脳循環障害により発生する可能性も示唆されていた。どちらも臨床的には、脳性麻痺(多くは半身麻痺)、てんかんおよび精神遅滞を引き起こす重篤な疾患だ。
研究グループは、IV型コラーゲンα1鎖をコードするCOL4A1変異が孔脳症を起こすことが報告されていたことから、日本人孔脳症患者61例および裂脳症患者10例の計71例でCOL4A1の変異解析を行い、孔脳症の10例と裂脳症の5例においてCOL4A1遺伝子のヘテロ接合性変異を同定した(画像1)。
その内の5変異については両親に認められない新生突然変異であり、2変異については明らかな臨床所見を認めない両親由来の変異だった。また、変異の見つかった1例は家族例であり、脳性麻痺を有する3名の患者と明らかな臨床所見を認めない血縁者にも変異が発見されている。

|
|
画像1。15の症例の詳細の一覧表 |
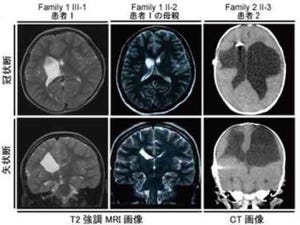
変異を有する患者の頭部MRI画像では片側性あるいは両側性の孔脳症や裂脳症が認められ、その程度もさまざまだ(画像2)。石灰化を伴うような「TORCH症候群」を疑う症例においてもCOL4A1変異が関与していることも判明している(画像2)。また、脳での異常(孔脳症・裂脳症)以外にも、目や筋肉の異常および溶血性貧血など幅広い表現型を引き起こすことも明らかとなった(画像1)。

|
|
画像2。COL4A1変異を有する患者の脳画像。患者1(Pt1)および患者2のCT画像では、側脳室周囲の石灰化を認め、TORCH症候群が疑われた。患者3、9、15(矢印)はそれぞれ裂脳症を呈している。患者7では片側の孔脳症が認められた |
今回の成果は、世界で初めてCOL4A1遺伝子変異が裂脳症を引き起こすことを明らかにした形だ。孔脳症・裂脳症は同じく血管障害によって引き起こされることも究明した。また、従来周産期障害が原因と考えられていた孔脳症の背景に、遺伝的要因が大きく関与していることも明らかにした形だ。
COL4A1変異が引き起こす幅広い表現型を明らかとしたことで、遺伝子変異を考慮すべき対象症例がわかってきたという。今後、COL4A1変異が見つかった方の脳出血予防法などの、変異に基づいた治療・管理法の確立が期待されるとした。