九州工業大学(九工大)は6月25日、独自に開発した深層学習モデルを用いて、タンパク質のアミノ酸配列情報のみで、98%という高い正答率で「ヒトとウイルス間のタンパク質相互作用」(Human-Virus Protein-Protein Interaction:HV-PPI)を予測することに成功したと発表した。
同成果は、九工大大学院 情報工学部 生命情報工学科の築山翔大学院生、同・倉田博之教授らの研究チームによるもの。詳細は、ライフサイエンスを題材とした学術誌「Briefings in Bioinformatics」にオンライン掲載された。
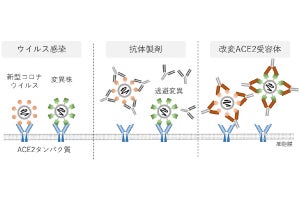
新型コロナウイルス(SARS-CoV-2)のヒト細胞への侵入は、ウイルス表面にあるスパイクタンパク質を、ヒト細胞の表面にある受容体タンパク質ACE2に結合させることから始まる。ここでポイントとなるのが、ヒトとウイルスの間で、両者のタンパク質が相互作用してしまうという点で、ヒト・ウイルス間タンパク質相互作用(HV-PPI)を同定することは、ウイルスの感染メカニズムを解明して、抗ウイルス薬(HV-PPI阻害剤)の標的を発見することにつながるとされている。
しかし、HV-PPIを同定するためには、果てしないトライ&エラーを重ねる必要があり、生物学実験では膨大な時間と費用がかかってしまい、現実的ではない。そこで期待されているのが、コンピュータによるHV-PPIの予測だという。
これまでのHV-PPIに関するコンピュータ予測法は、タンパク質立体構造情報に基づく分子動力学法を用いたドッキングシミュレーションであった。
-
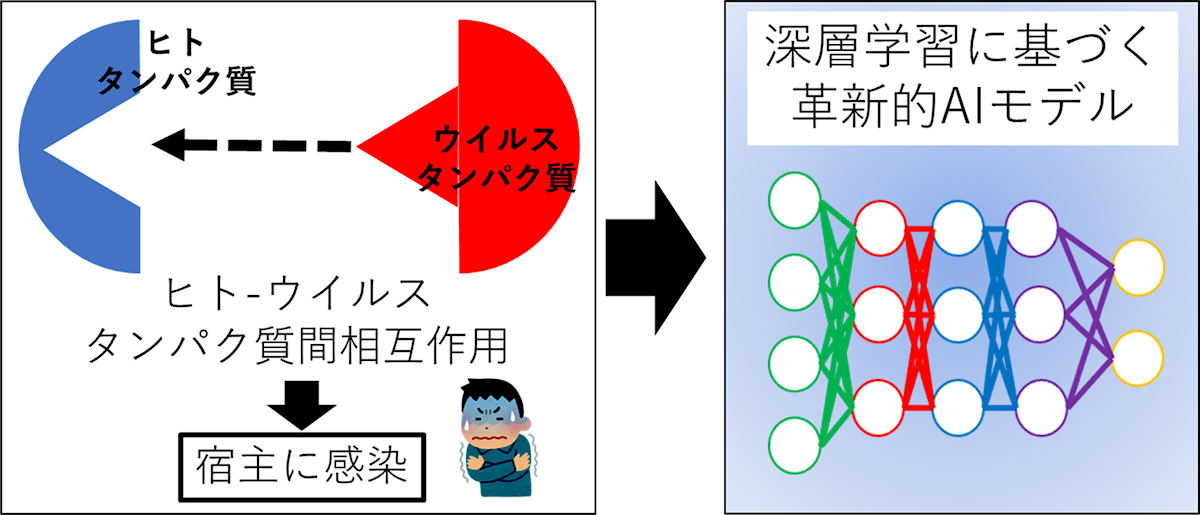
ヒトとウイルスのタンパク質間相互作用を予測する深層学習モデルの開発の模式図 (出所:九工大プレスリリースPDF)

しかし立体構造情報を解明することは難しく、ウイルスやヒトのタンパク質立体構造は不明なものも数多くあるため、分子動力学法のHV-PPI予測への応用は限定的となっていたという。
一方、タンパク質立体構造情報を用いることなく、ゲノムの塩基配列情報(タンパク質のアミノ酸配列情報)のみからHV-PPIを予測できれば高速化が可能とされるが、立体的構造を考慮せずにアミノ酸配列情報だけを用いるHV-PPI予測法は困難な課題だとされていたという。
そこで研究チームは今回、タンパク質のアミノ酸配列を文脈の流れと捉え、長鎖アミノ酸配列の順序パターンから鍵と鍵穴の立体的特徴を抽出することで、この問題を克服できると考察。そこで、このアイデアを実装するために、時系列データから未来を予測する深層学習モデル「Long Short-Term Memory」(LSTM)と、自然言語における単語の分散表現技術「word2vec」を応用した研究を実施したという。
その結果、LSTMとword2vecの両者を組み合わせて開発した深層学習モデルは、アミノ酸配列の文脈の流れから鍵と鍵穴の立体的特徴を的確に捉えて、正答率98%という高いレベルでHV-PPIを予測することに成功したという。今回開発された深層学習モデルは「LSTM-PHV」と命名され、専用Webサイトにおいて一般にも公開されているという。
なお、LSTM-PHVは、タンパク質のアミノ酸配列情報があれば、SARS-CoV-2を含む多様なウイルス群や変異型ウイルス群とヒトのHV-PPIの予測を可能だという。HV-PPIの正確な予測は、ウイルスの感染メカニズムを解明して、抗ウイルス薬の開発に貢献することが期待されている。