東京大学は12月19日、科学技術振興機構・戦略的研究推進事業(CREST)研究において、理化学研究所(理研)との共同研究により、「筋萎縮性側索硬化症(ALS)」の原因メカニズムを明らかにしたと発表した。
成果は、国際医療福祉大学 臨床医学研究センターの郭伸特任教授(東京大学大学院医学系研究科 疾患生命工学センター 臨床医工学部門 客員研究員)、東大大学院 医学系研究科 疾患生命工学センター 臨床医工学部門の山下雄也特任研究員、理研の西道隆臣チームリーダーらの共同研究グループによるもの。研究の詳細な内容は、12月18日付けで英国オンライン専門誌「Nature Communications」に掲載された。
ALSは筋肉を動かす運動ニューロンの変性・死滅が、呼吸機能も含む進行性の筋力低下を引き起こすという、主に初老期以降に罹患し、発症から数年の内に死に至る難病だ。
患者数は日本だけでも8000人を超え、加齢と共に頻度が増し、60歳以降の罹患危険率は300人に1人ともいわれており、決して稀な難病ではない。そして病因不明のため、有効な治療法が現時点ではない。
これまでの研究で、病因に関わる遺伝子やALSに特異的に見られる分子異常は特定されてきたが、未だその因果関係や運動ニューロン死に至るまでのメカニズムが解明されておらず、病因判明には至っていなかった。
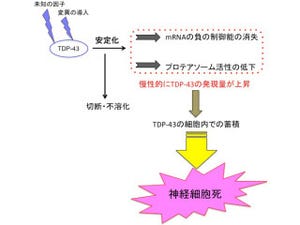
研究グループはすでにこれまでの研究で、ALSの病因に関わる疾患特異的分子異常として異常な「カルシウム透過性AMPA受容体」が発現していることを発見しており、今回の研究により、この異常が「カルパイン」の活性化を通じてもう1つの疾患特異的分子異常である「TDP-43」病理を引き起こしているという分子連関を解明することに成功した。
これにより、これまで知られていたALSの病因に関わる2つの分子異常のメカニズムと分子関連が明らかにされたこととなる。特にこの成果は、ALS患者の大多数を占める、遺伝性のない孤発性ALSの病因を説明するメカニズムであり、治療へ向け1歩前進したといえるだろう。
また、研究グループは、これまでの研究により、ALSでは、神経伝達に関わる「グルタミン酸受容体」のサブタイプである「AMPA受容体」に生じている異常にカルシウムを透過する分子変化が運動ニューロン死の原因であることを突き止めている。すなわち、AMPA受容体のカルシウム透過性を規定するサブユニットである「GluA2」に、本来生ずべきRNA編集(転写後の一塩基置換)が起こらず、未編集型GluA2が発現するために、カルシウム透過性が異常に高いAMPA受容体がALSの運動ニューロンに発現していること、これがRNA編集酵素である「ADAR2」の発現低下のためであることが確かめられていたのである。
この分子異常はALSに疾患特異的であるばかりではなく、ADAR2のコンディショナルノックアウトマウス(AR2マウス)の解析から運動ニューロン死の直接原因であることが判明し、この分子異常がALSに病因的意義を持つことが示されてきた。他方、ALSの運動ニューロンではTDP-43タンパクが断片化し、正常な局在である細胞核から喪失すると共に、細胞質に異常な封入体を形成することが生化学的・病理学的観察から明らかになっており、しかも、TDP-43の局在異常(TDP-43病理)とADAR2の発現低下はALSの同じ運動ニューロンに共存することも明らかとなり、両者の間には分子連関があると想定されていたが、両者の間にどのような分子連関があるのか、何故このようなTDP-43病理がALSの運動ニューロンに現れるのかについては不明だったのである。
今回の成果は、ADAR2発現低下が引き起こすTDP-43病理の形成メカニズムを明らかにしたもので、多くの研究者が抱いていた疑問を解明し、病因理解を1歩進めるものとなる。
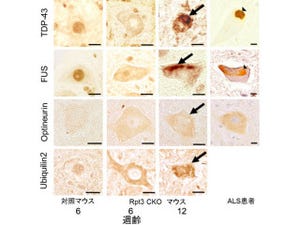
研究グループが開発したALSの分子病態モデルマウスであるAR2マウスで、TDP-43が正常な局在である細胞核から細胞質に移動し凝集塊を形成する病理変化が見られることが明らかとなった(画像1)。
画像1は、AR2マウスの脊髄前角運動ニューロンでのTDP-43免疫組織化学的観察。正常の運動ニューロンでは、TDP-43は、核に局在するが(画像1A)、AR2マウスの運動ニューロンでは、細胞質に封入体(inclusion)が観察され、核のTDP-43の染色性が低下(画像1B 矢印)ないし消失(画像1B 星印)する。TDP-43(緑)、TOPRO-3(青:細胞マーカー)。画像1Cは、ALS患者脊髄運動ニューロンのTDP-43病理。核のTDP-43が消失し細胞質に封入体が形成されているのが分かる。

|
|
画像1。AR2マウスの脊髄前角運動ニューロンでのTDP-43免疫組織化学的観察 |
さらに、この病理変化が生じるにはカルシウム依存性プロテアーゼであるカルパインによる「易凝集性断片」への切断が特異的に関わることを培養細胞系で証明し、その切断点を質量分析計を用いて同定。AR2マウスに見られたTDP-43病理がカルパインによるTDP-43切断に依ることを証明するために、カルパインが活性化していること、カルシウム透過性AMPA受容体の発現阻止やカルパインの内因性阻害物質である「カルパスタチン」の過剰発現ではTDP-43病理が起こらないこと、カルパスタチンのノックアウトマウスでは逆に増強することが示された。
さらに、ALS患者の剖検脳脊髄を用いて、患者でもカルパインが活性化し、カルパイン特異的TDP-43断片が発現していることも突き止め、TDP-43遺伝子の変異によるALSのTDP-43病理には、変異によりTDP-43がカルパインの切断を受けやすくなることが原因であることを明らかにした。これによりTDP-43病理は、ALS以外の前頭側頭葉変性症、アルツハイマー病などにも観察されるが、カルパインが関与している可能性が示されたこととなった。
未解明であったALSのTDP-43病理形成メカニズム(画像2)が明らかにされたことで、今後、ALSの病因の理解が進展することが期待される。また今回の成果は、ALS以外の前頭側頭葉変性症、アルツハイマー病などの神経疾患に観察されるTDP-43病理全般に通じることが示されたことから、TDP-43病理を呈する神経変性疾患に共通した病因メカニズムを理解する上でも役立つ知見となることが期待される。

|
|
画像2は、ADAR発現低下がTDP-43病理を形成するカスケード。Aが正常な運動ニューロン。ADAR2により編集型GluA2(GluA2R)が発現し、シナプスに発現するAMPA受容体のCa2+透過性は低い。B/C/DはALS運動ニューロンでADAR2発現低下がTDP-43病理を形成する以下の(1)から(5)のカスケードを示す。 |
なお、今回の成果により、ALSの病因メカニズムの解明が進むのと同時に、ALSの特異的治療の標的がさらに絞られることとなり、ALSの治療法が開発される可能性が現実的になってきたこととなる。そのため研究グループでは今後、明らかになった分子異常を標的とした治療研究を進めると共に、分子カスケードの上流下流をさらに検索していく予定としている。