理化学研究所(理研)は、京都府立大学、岡山県農林水産総合センター、京都大学、独マックスプランク研究所の協力を得て、DNA配列解析装置の次世代シーケンサーを用いて、「イチゴ炭疽病菌」と「ウリ類炭疽病菌」の全ゲノムを解読し、病原性に関与する遺伝子群候補を同定したと発表した。
成果は、理研植物科学研究センター 植物免疫研究グループの白須賢グループディレクター、パメラ・ガン特別研究員と、京都府大 生命環境科学研究科の久保康之教授、京大 農学研究科の高野義孝准教授、同・入枝泰樹特定研究員、同・池田恭子技術補佐員、岡山県農林水産総合センター 生物科学研究所の鳴坂義弘専門研究員、同・鳴坂真理流動研究員、独マックスプランク研究所のリチャード・オコネルグループリーダーらの国際共同研究グループによるもの。研究の詳細な内容は、12月14日付けで英国科学誌「New Phytologist」オンライン版に掲載された。
植物病害の8割前後が「植物病原糸状菌(カビ)」によるといわれている。植物病原糸状菌がもたらす農作物への病害に対抗し、環境に優しく安定した収穫ができる保護戦略の確立が、生産性向上や食の安全性確保のために重要だ。
植物病原糸状菌には、一般に1000種以上の分泌タンパク質をコードする遺伝子がゲノム上にあるという。植物病原糸状菌は、これらの膨大な分泌タンパク質を使って植物組織を破壊し、植物が本来持っている生体防御システムを妨害して感染すると考えられている。
従って、植物病原糸状菌が感染した時の分泌タンパク質の同定や発病機構を理解することは、新しい保護戦略を開発していく上で非常に重要だ。しかし、これまで植物病原糸状菌について詳しく解明されていなかった。
植物病害の中で特に大きな被害をもたらすのは「炭疽病」だ。炭疽病は、約40種ある「コレトトリカム(Colletotrichum)属菌」によって引き起こされ、それらによる病害は、600種以上の穀物、野菜、果樹、花卉(かき)などで確認されている。
被害は世界各地に広がり、日本ではイチゴ炭疽病菌(画像1)とウリ類炭疽病菌による被害が特に多く、イチゴ炭疽病菌の年間被害金額は約35億円とされ、その防除対策の確立は急務だ。そこで研究グループは、イチゴ炭疽病菌とウリ類炭疽病菌のゲノム解析を行い、病原性に関与する遺伝子の同定に挑んだのである。
画像1は、イチゴ炭疽病。A:イチゴの葉、B:イチゴの実、C:イチゴの葉の走査型電子顕微鏡(SEM)写真。炭素病菌により、長径3~7mmの病斑が葉や果実に発生し、この病斑が拡大すると茎や葉柄をとりまき、折れたりして枯れる。

|
|
画像1。イチゴ炭疽病 |
研究グループは、炭疽病の病原性発現に重要な遺伝子を同定するため、イチゴ炭疽病菌とウリ類炭疽病菌の全ゲノム解析が次世代シーケンサーを用いて行われた。そして得られた情報はバイオインフォマティクス(生物情報科学)により統合され、遺伝子の位置と機能情報を付加することが試みられたのである。
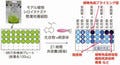
その結果、ウリ類炭疽病菌のゲノムの全長平均長は88.3Mb(1Mb:106塩基対)で、遺伝子総数は1万3484個だった。一方のイチゴ炭疽病菌のゲノムの全長平均長は56.2Mbで、遺伝子総数は1万5469個という結果となったのである(画像2)。
すでに報告されているイネ科炭疽病菌、アブラナ科炭疽病菌のゲノムの全長平均長が、それぞれ51.6Mbと49.1Mbであることから、ウリ類炭疽病菌のゲノムが特別に大きく、その原因は「AT-rich配列」が過剰に増加したためあるとわかった。
画像2。炭疽病菌のゲノム解析データ。A(アデニン)とT(チミン)の割合が非常に高いAT-rich配列の過剰な増加により、ウリ類炭疽病菌のゲノムの全長平均長はほかの炭疽病菌に比べて大きい。スキャフォールド:ゲノム配列断片の部分的な類似性を基に結合し、長い配列として再構成により構築された長い塩基配列。スキャフォールド数はその総数。

|
|
画像2。炭疽病菌のゲノム解析データ。スキャフォールドとは、ゲノム配列断片の部分的な類似性を基に結合し、長い配列として再構成により構築された長い塩基配列。スキャフォールド数はその総数 |
さらに、ゲノム上に存在する遺伝子をより詳細に調べるため、感染時のイチゴ炭疽病菌とウリ類炭疽病菌からタンパク質をコードするメッセンジャーRNA(mRNA)を抽出し、相補DNA(cDNA)を作成して次世代シーケンサーで解析を進め、遺伝子リストを作成。この遺伝子リストを基に、「カスタムマイクロアレイ」をデザインし、これを用いて感染時に特異的に発現する遺伝子群が同定された。
次に「qRT-PCR法」を用いてこれらの遺伝子群の発現定量解析を実施。その結果をほかのすでに報告されている2つの炭疽病菌(イネ科炭疽病菌、アブラナ科炭疽病菌)の遺伝子リストと比較し、炭疽病菌に共通する遺伝子群が同定されたのである。
これらの遺伝子群の特徴は、毒素などの低分子化合物を生産するための二次代謝酵素群やタンパク質を分解する加水分解酵素などをコードする遺伝子群などが多いことが判明した。
特にイチゴ炭疽病菌ではその数が多く、さらに炭水化物結合タンパク質をコードする遺伝子についてはウリ類炭疽病菌の約2倍の127個も保有しており、このイチゴ炭疽病菌では植物細胞壁の分解・形成するシステムが機能的に働くことを示唆している。
研究グループは、さらにほかの細菌や植物などのゲノムデータベースを利用して似た遺伝子群を検索。すると、炭疽病菌は遺伝子の水平伝播によって獲得した遺伝子群があることが見出された。
特にイチゴ炭疽病菌は、植物の成長を促す植物ホルモンのオーキシンの合成酵素をコードする遺伝子群をほかの細菌から獲得し、植物の成長に関与する機能を持つとわかったのである。
次に、以上のゲノム情報を用いて分泌タンパク質発現解析を行い、病原性に関与するタンパク質の候補を同定。また、それらが感染時前半(1日以内)と感染時後半(3日以後)で発現が異なることが見出され、その発現する遺伝子群がそれぞれ同定された。具体的には、病原性に関与するタンパク質候補群の内、感染時前半に130個の遺伝子の発現が10倍以上に上昇することがわかったのである(画像3)。
また、感染時前半には二次代謝酵素の発現が高いこと、それに対し加水分解酵素などは、感染時後半に主に発現することも明らかになった。これにより、炭疽病菌は異なる時期でそれぞれに特異的な遺伝子群を発現して、2つの感染ステージを形成していることがわかったのである。
画像3は、病原性分泌タンパク質候補をコードする遺伝子の発現パターン。試験管内で培養したもの(VH)、炭疽病接種前の胞子(C)、接種後1、3、7日後(dpi)の葉における、病原性分泌タンパク質候補の発現量の比較。赤色が発現のレベルが高く、緑色が低い。

|
|
画像3。病原性分泌タンパク質候補をコードする遺伝子の発現パターン |
今回の成果により、DNA鑑定による正確な炭疽病診断ができるようになり、さらに農薬耐性菌の選別などが可能になるという。また、病原性に関与する遺伝子群候補の同定により、これをターゲットする農薬の開発やこれらの遺伝子がコードする病原性タンパク質を認識して抵抗性を持つウリ科作物やイチゴなどの選定などの品種改良に貢献すると期待できるとしている。